

物质科学的核心问题之一是理解原子间的相互作用。如果我们能建立一个原子尺度的通用力场,便可创造一个原子间相互作用的求解器,从而通过分子动力学模拟诸多物质科学问题。前辈科学家们发明的量子化学/密度泛函理论(Quantum Chemistry/Density Functional Theory)通过求解薛定谔方程造就了一个模拟物质科学的大一统方法。通过此类方法,人们可以精确求解任意化合物体系中原子间相互作用,较为完美的解决了这个问题。但是密度泛函理论需要强大的算力,仅适用于数十至数百原子体系,无法线性扩展至大体系或复杂体系;而传统的分子动力学方法,虽然运算速度快,但是力场精度低、力场适用体系少,因此存在诸多限制。
中国科学院物理研究所/北京凝聚态物理国家研究中心孟胜、刘淼研究员联合松山湖材料实验室团队,研发了一种基于深度学习图结构的通用预训练力场(graph-based pre-trained transformer force field ,简称GPTFF)。GPTFF模型充分利用Atomly数据库中的海量数据和 transformer 算法的注意力机制,可精确预测原子间相互作用,具备出色的精度和泛化能力,适用于几乎任意无机化合物的近平衡态,可用于大体系及复杂体系的分子动力学模拟。GPTFF模型支持开箱即用,用户可跳过模型训练步骤,直接开展化合物的结构优化、相变模拟、物质输运等科学问题。GPTFF开发团队已将该模型的算法、程序、模型参数开源,所有人都可下载使用。
原子尺度的通用力场AI大模型,可广泛支持物质物理、材料、化学等诸多领域,它也被认为是物质科学的基座模型,有望变革性地改变物质领域的计算模拟方式。美国的 Materials Project团队率先发布了m3gnet和CHGNET模型及数据集,成为了该方向的标杆。google、微软等科技公司洞悉到该方向的重要性,也正在研发类似的AI模型及数据产品。我国虽然有很多类似的项目及模型,但都采用欧美的开源数据集,无法从根本上实现自主可控,更无法实现超越。GPTFF模型的训练数据源自自研的Atomly材料数据库(https://atomly.net/)。该数据库包含近35万个无机材料数据,数据规模和质量位于世界顶级水准,知识产权自主可控,也是我国唯一可对标欧美竞品的无机材料数据库。GPTFF的训练数据包含3780 万个单点能量、117 亿个力对和 3.402 亿个应力,数据具有高度的标准和和一致性,保证了模型的高精度和强泛化能力。GPTFF能够实现对原子间相互作用的精确预测,原子间能量、受力以及应力的预测精度分别达到 MAE=32meV/ 原子、71 meV/Å 和 0.365 GPa,优于m3gnet和CHGNET(图1)。可用于模拟晶体结构弛豫、固态电解质中的离子疏运、金属在应力下的相变等科学问题 (图2)。
本工作受到了国家自然科学基金项目、科技部重点研发计划和中国科学院的资助。物理所博士生谢帆恺是该论文第一作者,孟胜研究员、刘淼研究员为通讯作者。该成果以“GPTFF:一套高精度开箱即用的无机化合物人工智能通用力场模型(GPTFF: A high-accuracy out-of-the-box universal AI force field for arbitrary inorganic materials)”为题发表在《Science Bulletin》杂志。
文章链接:
https://doi.org/10.1016/j.scib.2024.08.039
模型开源代码、参数下载链接:
https://github.com/atomly-materials-research-lab/GPTFF
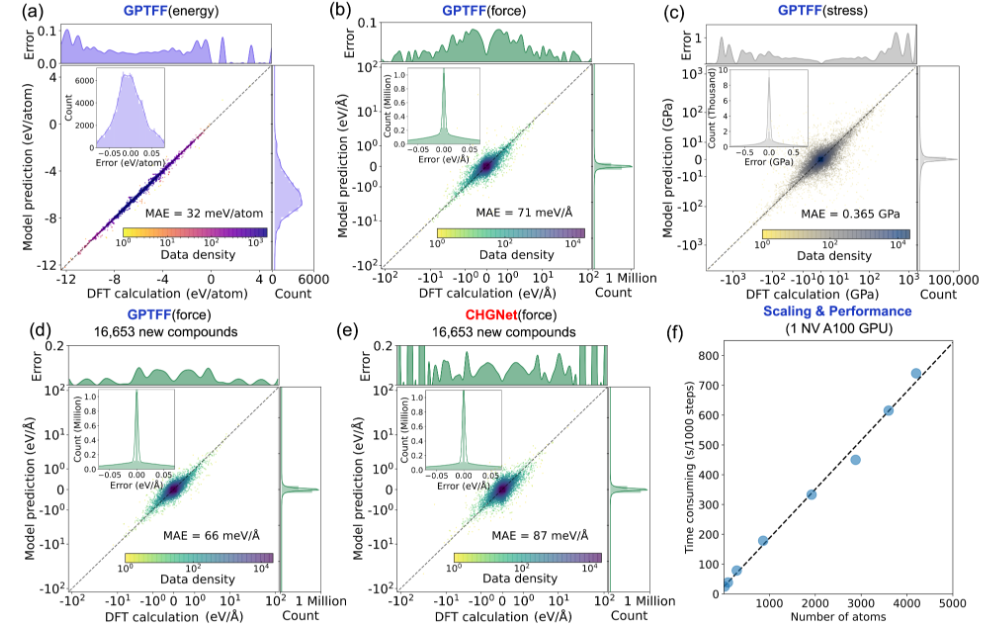
图1. 模型精度及运行效率
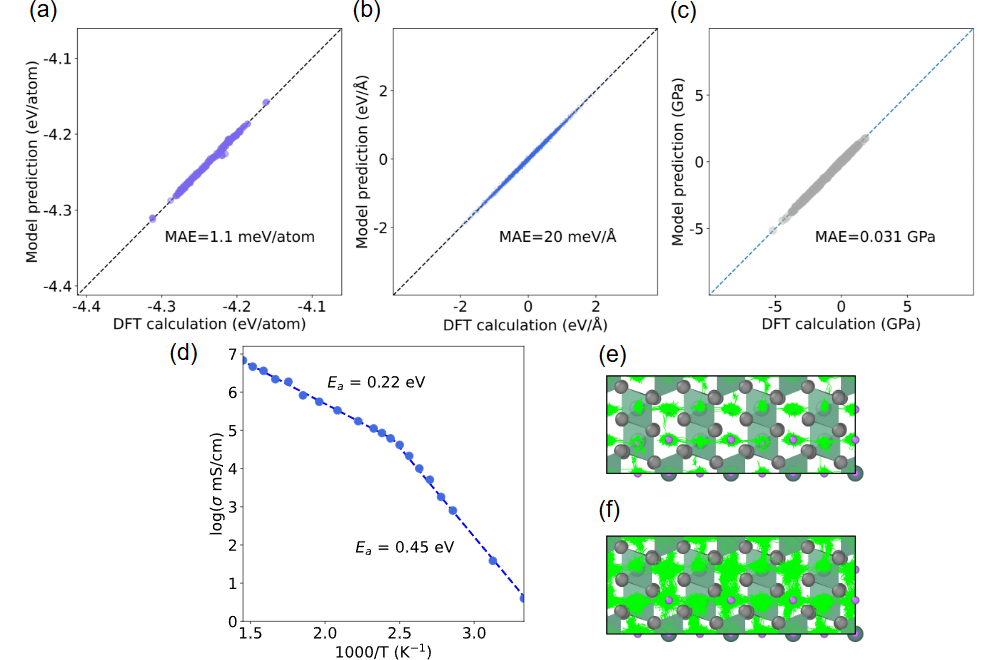
图2. 模拟锂离子电池固态电解质中的Li离子输运行为