

在一定温度下,晶态材料中电子自旋之间的相互作用会诱导出铁磁和反铁磁等多种长程磁有序态,磁矩一般来自于原子3d/4f 壳层中的未成对电子,也可以来自于一些缺陷和p电子。p电子诱导的磁性很早就有报道,典型的例子是氧气在低温下所形成的固态氧,它表现出长程的反铁磁性(TN=24 K),这里的磁矩来源于氧气分子中未成对的π*(pxy)电子。近年来,人们发现在一些化合物中分子氧作为阴离子(O2-和O22-),可以诱导出丰富的磁性,如AO2 (A=Na, K, Rb, Cs)中的反铁磁相变和Rb4O6/Cs4O6的短程自旋玻璃。同时,分子氧在一些化合物中也可以作为阳离子(O2+)并诱导磁性,如O2PtF6的亚铁磁性。对于长程铁磁性,实验上缺乏可靠的证据,虽在RbO1.72和KxBa1-xO2 (x=0.269)中观察到了微弱的铁磁信号,但都没有表现出本征的长程铁磁序,目前对长程铁磁序形成机理的认识仍然很有限。
中国科学院物理研究所/北京凝聚态物理国家研究中心先进材料与结构分析实验室A02组博士生邓俊、郭建刚副研究员和陈小龙研究员分析了AO2 (A= K, Rb, Cs)中O2-二聚体的磁性相互作用规律,提出了改变化合物中层与层之间O2-二聚体的排布方式来调控磁性,从而诱导长程铁磁序的学术思想,如图1所示。利用第一性原理计算结合结构搜索的方法,他们筛选出了具有铁磁性的三元半金属(half-metal)化合物α-BaNaO4,空间群是P4/mmm,其中所有的O2二聚体朝着c方向,并且层与层之间的二聚体以头对头的方式排列,如图2所示。分子动力学、形成能和声子谱等分析证明它在500 K下是稳定的。电荷分析显示,化合物中O2的平均价态为-1.5, 故未完全填满的π*(pxy)轨道呈现磁矩。对可能的磁结构进行能量计算后,发现铁磁态的能量最低,即它的基态是铁磁态。对其电子结构计算表明,α-BaNaO4是具有4 eV自旋能隙的半金属,如图3所示。平均场理论估算其居里温度Tc为120 K。本工作首次预言了一个含O2二聚体的半金属氧化物,正是这种独特的排列方式实现了层与层之间分子磁矩的直接交换作用,最终诱导出长程铁磁序。这些结果加深了对O2分子诱导铁磁性机理的认识,为设计p电子诱导的磁性材料提供了新的思路,以上成果发表在J. Am. Chem. Soc. 142, 5234-5240 (2020)。同时,国内外已有的工作表明这类化合物还具有其它重要的物理性质,如压力引起的电荷序、强的电子关联、Mott相变等,值得进一步深入研究。
该研究组在前期的工作中,曾预测了含N2二聚体的半金属化合物MN4 (M=Mn, Fe, Co)。它们具有高的居里温度(~103 K)和大的自旋带隙(~5 eV,HSE06),其中,大的自旋带隙来源于N=N双键中π*和π轨道之间较大的能量差。与α-BaNaO4不同的是,它的磁矩来源于过渡金属而非N2二聚体。同时,N2二聚体还能和主族元素形成化合物SiN4,表现出与金属Cu相比拟的电导率和热导率。这些结果表明了N=N双键也可以作为设计磁性、高热导与高电导材料的结构基元,为探索功能材料提供了新的思路,相关成果发表于Phys. Rev. B 99, 184409 (2019)和Chin. Phys. Lett. 35, 087102 (2018) 。
上述工作得到了科技部战略性国际科技创新合作重点专项(2018YFE0202601),国家自然科学基金委(51922105, 51532010, 51772322,91422303), 国家重点研发计划(2016YFA0300600,2017YFA0304700), 中国科学院前沿科学重点项目(QYZDJ-SSW-SLH013),北京市科学技术委员会(Z161100002116018)和中科院B类先导专项 (XDB07020100)等项目的支持。
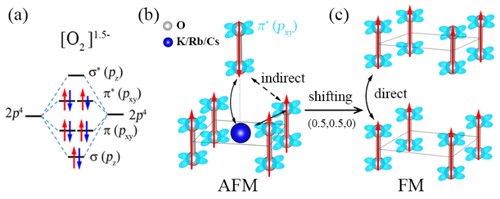 |
| 图1. (a) 化合物α-BaNaO4中O2二聚体的轨道排布和电子占据示意图。(b)AO2(A=K, Rb, Cs)中O2二聚体排列方式以及间接交换示意图。(c) 改变O2二聚体排列方式后的直接交换作用示意图。 |
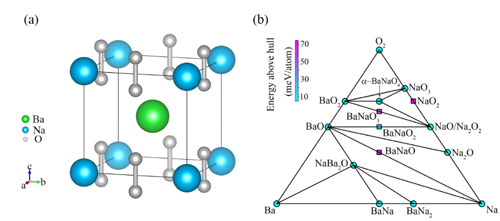 |
| 图2. (a) 化合物α-BaNaO4的晶体结构。(b) 基于形成能构建的Ba-Na-O三元相图。 |
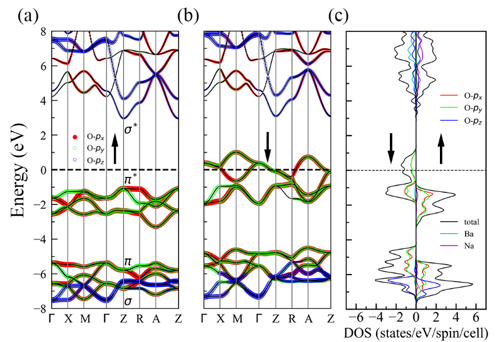 |
| 图3.α-BaNaO4中(a)自旋向上和(b)自旋向下的能带结构,不同颜色代表不同轨道电子对能带的贡献。(c)α-BaNaO4中两个自旋方向的分波态密度。 |