

金属离子的不同电荷态反映了最外层电子数目与轨道占据等情况的变化,特定的电荷有序分布从根本上决定了材料的晶体结构与电子性质。在PbMO3(M代表3d过渡金属)钙钛矿家族中,随着d电子数目的增加,Pb的价态由+2价(如PbTiO3、PbVO3)逐渐转变为+4价(如PbNiO3),而对于家族中间的成员比如PbCoO3,前期研究表明Pb2+与Pb4+这两种不同电荷态在A位形成1:3的有序分布,对该材料施加原位高压可诱导B位Co2+离子的自旋态改变以及A-B位间的电荷转移(参见:JACS, 139, 4574, 2017;142, 5731, 2020)。由于离子尺寸的失配,合成A位含有Pb4+的钙钛矿氧化物通常需要高压高温实验条件。作为与PbCoO3相邻的成员,PbFeO3因缺乏单相性良好的高质量样品,其电荷有序分布形式及与之关联的物理属性一直悬而未决。
近期,中国科学院物理研究所/北京凝聚态物理国家研究中心磁学国家重点实验室M08组龙有文研究员团队,利用高温高压技术获得了高质量PbFeO3钙钛矿材料,通过广泛合作发现了A位全新的电荷有序分布形式,并由此导致了B位Fe3+离子在高达418 K的临界温度发生自旋重取向转变,表明电荷有序是调控自旋取向的一种新方法。
经过大量尝试探索,团队成员寻找到了制备高质量PbFeO3多晶样品的最佳条件,合成压力必须高于8 GPa,且加热温度只能在1150℃附近(浮动不超过50℃)。为确定材料的晶体结构与综合物理性质,团队进行了同步辐射X光衍射、X射线吸收谱、高分辨电子衍射、磁化率、磁化强度、电输运、高低温中子衍射等系列实验测试以及基于第一性原理的理论计算。结果表明,PbFeO3的B位由单一的Fe3+离子组成,但A位由Pb2+与Pb4+这两种电荷态按1:1的比例组合而成。并且,Pb2+与Pb4+在A位形成全新的-A-B-B-型层状电荷有序,其中A层为单一的Pb2+,B层为75%的Pb4+与25%的Pb2+有序组合而成。这种新颖的电荷有序在其他化合物中未曾报导,使得材料形成2ap×6ap×2ap的超晶胞(ap代表简立方钙钛矿的晶格常数)。相应于A位特殊的电荷有序,B位Fe3+离子在A-B与B-B层间形成具有不同对称性的两种原子位置,导致FeO6八面体不同的各向异性晶体场。PbFeO3在600 K发生倾斜反铁磁自旋有序,自旋方向大致平行于a轴,宏观上表现出弱铁磁性。令人意外的是,该材料在高达418 K的临界温度自旋取向发生90°旋转,随着温度降低形成自旋平行于b轴的共线反铁磁结构。在以往的Fe基氧化物中,自旋重取向往往发生在室温以下,且与A位稀土离子或者B位磁性离子掺杂引入的磁各向异性相关。理论计算表明,PbFeO3钙钛矿中A位特殊的层状电荷有序导致了B位两种Fe位置磁各向异性能的竞争,其竞争结果是自旋重取向转变的主要原因。由此可见,电荷有序是有效调控自旋取向的一种新机理,PbFeO3远高于室温的自旋重取向行为有利于该效应在自旋电子学器件领域的可能应用。
相关研究结果发表在近期的Nature Communications上[Nature Communications 12, 1917 (2021)]。本研究工作获得了物理所靳常青研究员课题组(赵建发博士、于润泽副研究员等)、日本东京工业大学M. Azuma教授团队、瑞士PSI Michel Kenzelmann教授团队、德国马普研究所、美国橡树岭国家实验室等多家单位成员的广泛合作。该工作获得了国家自然科学基金委(11934017, 51772324, 11921004, 11904392)、科技部(2018YFE0103200,2018YFA0305700)、北京市自然科学基金(Z200007, 2202059)、中国科学院(XDB33010200, QYZDB–SSW–SLH013)等的支持。
文章链接:https://www.nature.com/articles/s41467-021-22064-9
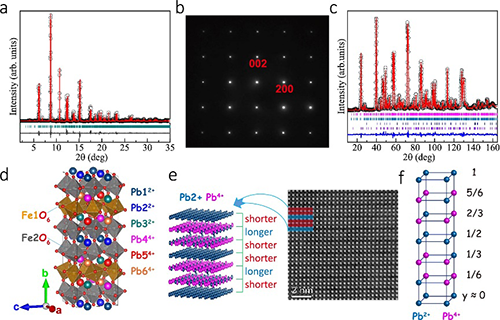
图1:PbFeO3的高压合成和晶体结构相关表征。
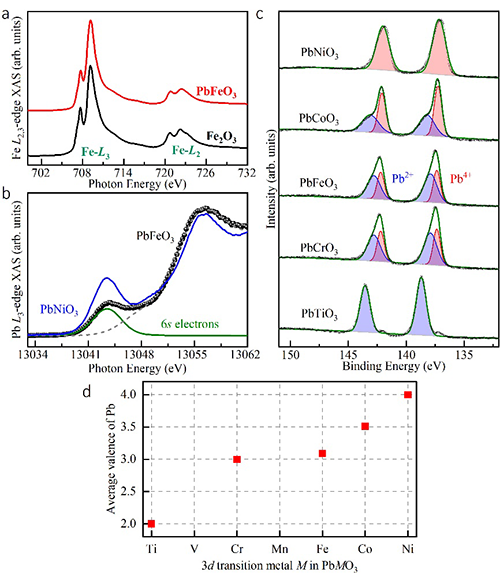
图2:PbFeO3 X射线吸收谱相关的价态表征。
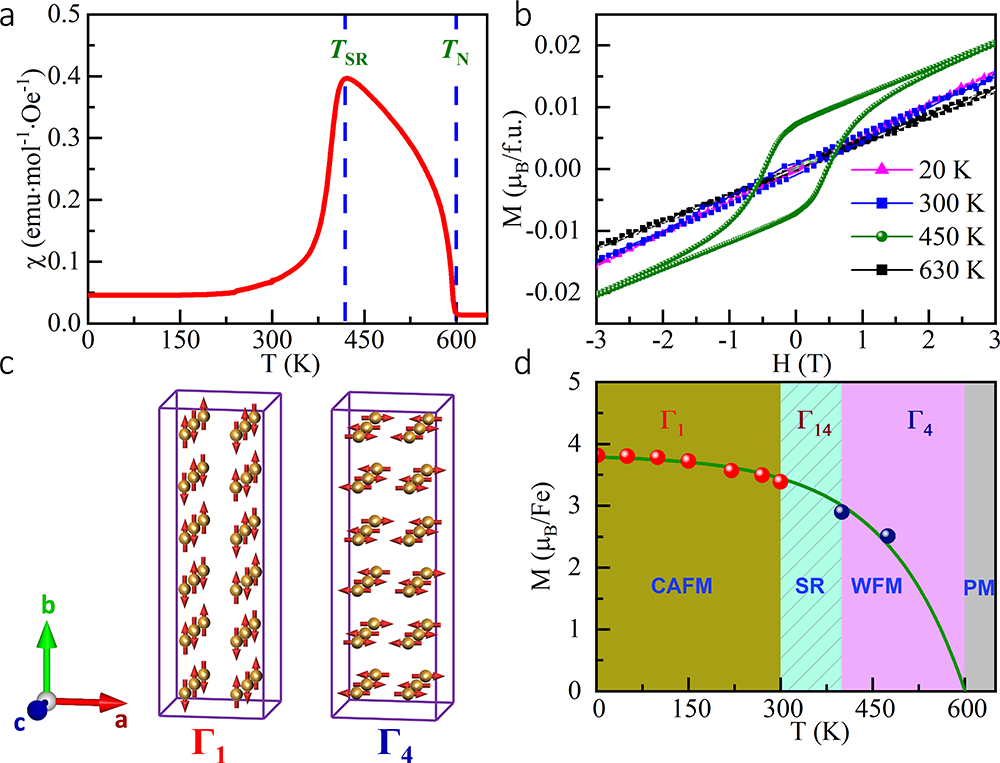
图3:PbFeO3磁化率、磁化强度、磁结构示意图及磁转变相图。