

Chevrel相化合物(Chevrel Phases,CPs)以其独特的AMo₆X₈(A为金属元素,X = S,Se,Te)团簇结构,在高场超导体、电池正极和电催化等领域展现出巨大潜力。其中,三元硒化物和碲化物CPs的合成极具挑战性,特别是对于碱金属这一类大离子半径插层的硒化Chevrel相,由于阳离子插层引发的晶格失稳导致其表现出热力学亚稳性,使得实验合成和物性研究长期受限。精准合成此类材料并揭示其超导特性,是探索高场超导材料体系CPs化合物及其应用的关键一步。
近日,中国科学院物理研究所/北京凝聚态物理国家研究中心超导国家重点实验室SC10组博士研究生石运清、任治安研究员以及重庆大学阮彬彬副教授,和先进材料与结构分析实验室的博士后马小平、杨槐馨研究员合作,在亚稳态钾插层硒化Chevrel相超导材料研究中取得重要进展。研究团队通过精确控制的低温固相反应,成功合成了化学式为K1+δMo6Se8(δ ∼ 0.37)的新型Chevrel相化合物,并观察到超导转变温度为8.9K、上临界场高达26.4T的体超导性(如图2所示),其上临界场显著超过泡利顺磁极限(μ₀HPauli≈16.6T),在高场应用研究方面表现出重要参考价值。
研究团队综合利用X射线衍射(XRD)和球差校正扫描透射电子显微镜(HAADF-STEM)技术,确认K1+δMo6Se8结晶于三斜晶系,空间群为P−1 (No. 2)(如图1a,b所示)。结构精修和原子尺度成像表明,钾阳离子主要占据Mo₆Se₈团簇间的六面体空腔(Site1),同时部分钾(约42%)占据了相邻团簇间的通道位置(Site 2),形成了δ ∼ 0.37的非化学计量比。这种钾离子部分占据通道位点(Site 2)的构型在碱金属插层的CPs中极为罕见。键价和(Bond Valence Sum,BVS)计算揭示了K-Se键存在极强的应力,特别是位于Site 2的钾,其键价和远高于理论值(K⁺),可能是导致其热力学亚稳性的主要原因。
这项工作不仅拓展了Chevrel相超导体系,特别是碱金属插层的硒化物,其突破泡利极限的特性为探索高场超导应用提供了新材料平台。K1+δMo6Se8中表现出的多带超导特征和高临界场,暗示了其中可能存在非常规的超导配对机制,有待未来更深入的光谱学研究(如扫描隧道谱、μ子自旋弛豫)揭示。物理所博士研究生石运清和博后马小平为共同第一作者。物理所任治安研究员和重庆大学阮彬彬副教授为共同通讯作者。该研究工作得到了国家重点研发计划、国家自然科学基金以及中国科学院先导专项的支持。该研究成果以“Superconductivity in Metastable K1+δMo6Se8: A Potassium-Intercalated Chevrel Phase”为题发表于Journal of the American Chemical Society,2025,147,22453–22459 (DOI: 10.1021/jacs.5c01401)。
文章链接: https://doi.org/10.1021/jacs.5c01401
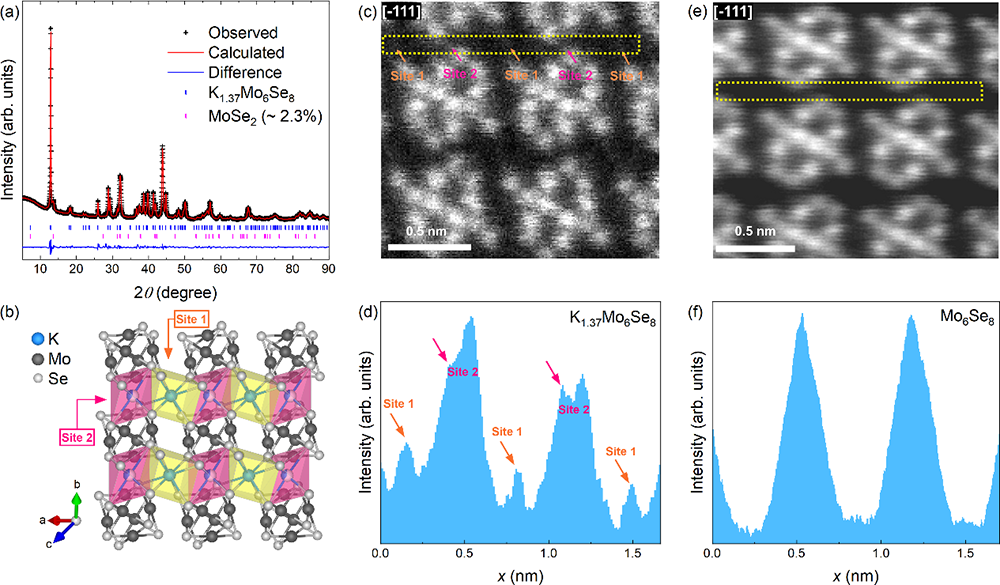
图1:K1+δMo6Se8的晶体结构表征。(a) XRD图谱及Rietveld结构精修结果。(b) 晶体结构示意图,显示Mo₆Se₈团簇及钾离子占据的Site1和Site2位置。(c,d) K1+δMo6Se8和(e,f) 母体Mo₆Se₈的HAADF-STEM图像及线性扫描,显示Mo、Se原子柱及钾离子位置。